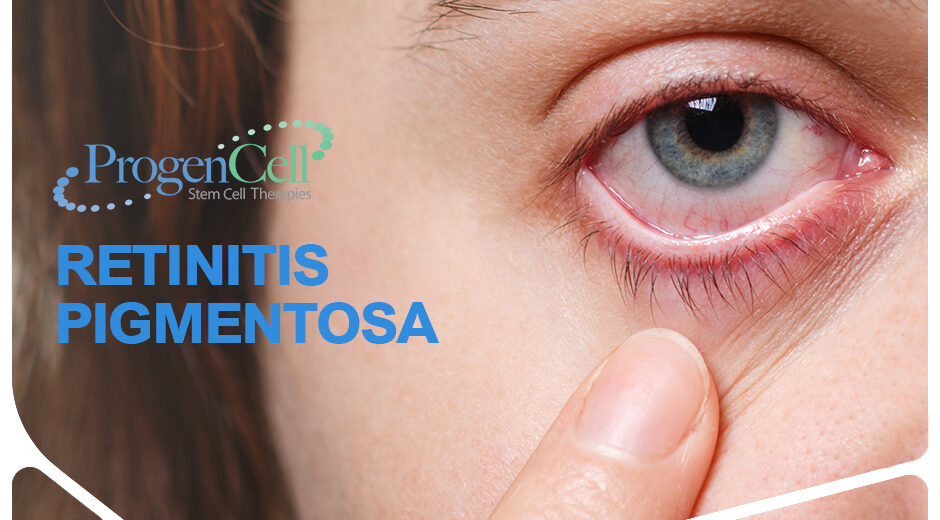

What is retinitis pigmentosa? is a progressive disease that affects the retina, affecting the peripheral vision with a slow evolution resulting on tunnel vision. Since there is not known cure, eventually it transforms into complete blindness.
Currently, traditional treatments for retinitis pigmentosa are palliative, meaning, they focus on improving the patient’s quality of life and reverse the cause of the disease therefore the patient has no option than accept the progress of the medical condition.
Retinitis pigmentosa is a group of hereditary origin genetic disorders that affect the retina, which is, the part of the eye that transforms light into nerve impulses. Therefore, patients with retinitis pigmentosa lose night vision in the early stages, and later lose peripheral vision, and then a gradual loss in their abilities to drive cars, read and recognize faces, even during the day.
People with retinitis pigmentosa often have other health problems that affect their vision, such as clouding of the lens (cataracts), glaucoma, nearsightedness, and retina inflammation, which accelerates vision loss. In addition, this disease is associated with neurological, metabolic or cardiac alterations, Usher, Barde-Biedl, Refsum and NARP syndromes (neuropathy, ataxia and retinitis pigmentosa), which entail extraocular symptoms.
Table of Contents
Symptoms of retinitis pigmentosa
, tunnel vision (below)")
- Night blindness. In addition to poor vision, there is a slow adaptation of the retina to darkness. This symptom usually occurs between the first and second decade of life.
- Tunnel vision. The reduction of the visual field in a progressive and concentric way is known as tunnel vision since central vision is preserved until the most advanced stages of the disease. This symptom makes it difficult for the patient to see objects on the periphery, and only allows vision focused on the center of the image.
- Photopsias. Glare and light perception in an anomalous way when there is a lot of light so the patient could see brightness, flashes or flashing lights.
- Cone dystrophy. Alteration of the color perception, a symptom that appears in advanced stages of the disease.
What is the main risk of retinitis pigmentosa?
Unfortunately, retinitis pigmentosa is a degenerative disease that produces a serious decrease in visual capacity that, in many cases, causes total blindness.
Retinitis pigmentosa is estimated to affect one in 4,000 people worldwide and is the leading cause of total blindness
Evolution of retinitis pigmentosa
- At what age does retinitis pigmentosa begins? The onset of the disease can occur between 10 and 30 years of age, even earlier, since it affects very young people, who begin to lose their vision at night, see flashing lights, flashes, or not recognize colors. The name “retinitis pigmentosa” is derived from this characteristic.
- Early stages of retinitis pigmentosa. Initially, there is a photoreceptors progressive loss , responsible for the perception of colors , light perception, sight to darkness adaptation and the retina pigment epithelium due to enzymes and protein mutations that compose it. Some peripheral vision quadrants also begin to be lost without any apparent cause.
- Subsequently, night blindness , central vision and progressive loss appear, keeping only the peripheral vision that, eventually, will also be lost gradually, starting with the decrease in skills to read, recognize faces, drive and perform other activities during the day.
Retinitis pigmentosa Treatment
There is currently no treatment to cure retinitis pigmentosa or recover lost vision. However, associated diseases, such as cataracts or inflammation of the retina can be treated, delaying the progression of the disease somewhat.
What vitamins are good for retinitis pigmentosa?
Dietary changes and vitamin supplementation can slow the progression of visual loss; the most recommended is vitamin A, or retinol, whose role is to form and maintain mucous membranes, skin, teeth and bones. In addition, it is an antioxidant that eliminates free radicals from the body, protecting DNA from mutations to slow down cellular aging.
This vitamin can be consumed in the form of carotenoid pigments or carotenes to prevent and fight respiratory, ocular and dermatological infections. It can also be found in foods such as egg yolk, liver, fish oil, seaweed and intense green color vegetables, as well as in yellow and orange fruits, or in the form of vitamin supplements.
Glasses for retinitis pigmentosa
Patients with retinitis pigmentosa are often advised to wear sunglasses with UV protection to avoid strong sunlight when going outside, and they can also wear them indoors in places with intense lighting.
In extreme cases, you can choose to take advantage of technological advances in artificial vision. Artificial vision devices are under development for patients with severe or total vision loss. These are special glasses that feature a small camera with a video processor, which converts images into stimulation patterns that send information to an implanted microchip in the patient’s retina.
Latest treatments for retinitis pigmentosa
Experimental treatments include gene therapy, retinal transplantation, and retinal prostheses. There are more than two hundred and sixty genes linked to retinitis pigmentosa, so genetic approach is very promising.
People suffering from this disease have wondered “Is there any surgery available for retinitis pigmentosa?”, the answer is yes, but not with much success. It is a retinal transplantation, which requires a recently deceased donor whose retina is compatible with the patient. Retinal prosthesis means to implant surgically. It’s the kind of mechanism that made a six-million-dollar man famous. However, so far, this treatment has not been very successful.
On the other hand, ophthalmologists specializing in ProgenCell have been applying autologous stem cells to this disease since 2008 and their results have been very promising. In most cases, the patient’s condition stops; best cases, the patients have shown very important recovery, to the extent that they were able to go back to their daily life activities with little limitation.
Steven, a patient from the United States, has allowed us to share his experience at ProgenCell. He was diagnosed with retinitis pigmentosa in November 2010. After two visits, he wrote to us:
“I went to three different doctors who said there is no treatment for my condition. So I kept looking on the internet and found ProgenCell. Some of my family and friends were against my decision, but we all knew it was the only chance to improve. The procedures were much faster than I expected and I felt no pain. I never had any side effects.”
“This is my second stem cell procedure and I have improved a lot. I can do things I used to do before. Now I have a better, clearer and broader vision. All my friends and family are excited about my progress, they all think it’s wonderful that this treatment is available.”
What causes Retinitis Pigmentosa?
Retinitis pigmentosa is caused due to the mutation of one or more genes responsible of forming the proteins necessary for visual photoreceptors to develop. Currently, more than a hundred of around 260 genes related to vision diseases have been detected, which are the cause of retinitis pigmentosa cause.
Precisely because they have a genetic origin, the use of stem cells has proven to be a very promising therapeutic alternative since they can promote the damaged tissues to regenerate and some stem cells could become tissue of the ocular area.
How is retinitis pigmentosa inherited?
Mutations related to this disease can be passed from parent to child through three types of inheritance:
- Autosomal dominant retinitis pigmentosa. There are 16 genes responsible for this type of retinitis pigmentosa; in the dominant type, one of the parents suffers from the disease and has a 50% chance of transmitting it to their children, however, it is one of the less severe forms since the symptoms appear later and progress slowly.
- Autosomal recessive retinitis pigmentosa. Three genes responsible for this type of retinosis have been detected: RHO, RP1 and RD5, as well as mutations in the protein beta-phosphodiesterase. In this type, one of the parents is a disease carrier but without having symptoms, it only has a 25% chance of transmitting the altered gene to one of their children; however, the symptoms are more severe.
- Retinitis pigmentosa linked to recessive sex. Six sex-related genes are known, of which mutations in the RPGR gene stand out. In this case, the cause is related to the X chromosome, so males are affected and females are carriers of the altered gene; that is, male parents do not pass on the disease to male children, but it is the mother who passes on the gene and affects males to a greater extent.
It is the most severe retinitis pigmentosa form as symptoms appear at an early age and evolve rapidly. This type of inheritance occurs in 10% of the families.
In addition to this, alterations in the same gene can cause other retinitis types, which is known as allelic heterogeneity. It is also important to note that people with the same mutation may have different symptoms, even if they are from the same family since other genetic and environmental factors influence.
On the other hand, there are retinitis pigmentosa cases without a family history, with the affected patient being the only family member with the disease due to a genetic mutation.
How to diagnose retinitis pigmentosa?
In order to make a retinitis pigmentosa differential diagnosis, the patient must be analyzed clinically, by examining his retina and applying visual tests to detect other associated diseases, such as astigmatism and myopia (linked to the X chromosome). Visual field vision, color vision, and patient adaptation to darkness or dim lights are also evaluated.
Some diagnosis test may be:
- Full-field electroretynography, which measures the electrical response of the retina to light.
- Campimetry to show peripheral and central vision.
- Multifocal electroretinogram, which allows many focal electroretinograms to be recorded at the same time to represent the the central cone function.
Timely diagnosis and specialized treatment are key to detecting associated diseases, delaying the retinitis pigmentosa symptoms and preserving the patient’s quality of life.
Stem cell treatment involves concentrating a large amount of stem cells in a very small volume. It is then infused in a retrobulbar form to release the cells near the optic nerve and behind the eyeball. The cells “bathe” the back of the eye and attach to begin their regenerative function.
Likewise, it is reinforced with an intravenous stem cells infusion, to increase the stem cells amount in the blood torrent and reach the veins that feed the eye and retina. The set of both applications makes the eye components regenerate and increases the vision effectiveness.
It is important to emphasize that the vision in retinitis pigmentosa is lost little by little. It degenerates tissues as time goes on. So the application of stem cells in early stages helps to stop or slow the disease progress. It is recommended to start the stem cells application upon getting the diagnosis to avoid reaching severe stages of the disease.
The more severe the case, the more difficult it is for the cells to repair damaged tissues, it is preferable to avoid degeneration.
If you or a loved one suffers from retinitis pigmentosa, begin recovery today by contacting ProgenCell for a free case evaluation and see our stem cell treatment for eyes in Mexico. We are specialists in other treatments as well, such as stem cell therapy for MS, stem cell therapy for parkinson’s, stem cells for psoriasis, for diabetes, stem cell treatment for macular degeneration, and more. The sooner you start, the better your results will be.






Facebook Comments